Part 1: Why Oxygen Is Critical — Why Transport Is a Problem
The Evolutionary Importance of Oxygen
The transition from anaerobic to aerobic life was one of the most pivotal events in the history of evolution. Oxygen dramatically increased the energetic yield of glucose oxidation:
- Anaerobic conditions: oxidation of glucose yields only 2 molecules of ATP
- Aerobic conditions: the same oxidation yields 38 molecules of ATP (2 from glycolysis + 36 from oxidative phosphorylation)
This 19-fold increase in energy efficiency allowed organisms to grow larger and more complex. However, larger organisms faced a new challenge: how to distribute oxygen efficiently to all of their cells.
Small organisms can absorb directly from the air or surrounding aqueous environment by simple diffusion. But for large, multicellular organisms, diffusion alone is far too slow and oxygen is too insoluble in water to meet metabolic demands. These organisms therefore evolved a circulatory system paired with specialized oxygen-carrying proteins.
The Chemical Problem
Oxygen presents two key challenges as a transported molecule:
- Poor aqueous solubility: has a maximum solubility of only M in water — far too little to supply metabolically active tissues.
- Limited diffusion range: Diffusion of oxygen through tissues is ineffective over distances greater than a few millimeters.
None of the standard amino acid side chains in proteins are chemically suited for reversible, specific oxygen binding. Nature’s solution was to incorporate transition metal ions — particularly iron () and copper () — which have a strong tendency to bind oxygen. However, free iron promotes the formation of highly reactive oxygen species (e.g., hydroxyl radicals) that can damage DNA and other macromolecules. Therefore, iron in biological systems is almost always sequestered in a tightly controlled environment: a heme prosthetic group.
Part 2: Protein–Ligand Interactions — Foundational Concepts
Before diving into specific proteins, it is essential to understand the general principles governing how proteins bind to small molecules.
Ligands and Binding Sites
A ligand is any molecule that reversibly binds to a protein. This includes small metabolites, ions, drugs, other proteins — essentially anything. The binding site is the region of the protein that accommodates the ligand. It is highly specific: complementary to the ligand in size, shape, charge, and hydrophobicity.
Key features of protein–ligand interactions:
- Binding is reversible and transient — this is critical for biological responsiveness.
- A protein may have multiple distinct binding sites for different ligands.
- Binding often triggers a conformational change (structural rearrangement), making the site even more complementary to the ligand. This is called induced fit.
- In multisubunit proteins, conformational changes in one subunit can propagate to other subunits — the basis of cooperativity and allostery.
Binding Sites vs. Protein Domains
These two terms are often confused. Here is a clear comparison:
| Feature | Binding Site | Domain |
|---|---|---|
| Size | Small — often just a few amino acids | Large — typically 50–300 amino acids |
| Structure | Part of the surface or interior | An independent folding unit |
| Function | Specific interaction with a ligand | Broader structural or functional role |
| Independence | Depends on the overall protein structure | Can fold and function independently |
| Relationship | Can be part of a domain | May contain multiple binding sites |
The Dissociation Constant ()
The reversible binding of a protein (P) to a ligand (L) is described by the equilibrium:
The dissociation constant is defined as:
To describe how much of the protein’s binding capacity is occupied at a given ligand concentration, we use the fractional saturation :
Key interpretations:
- When , then : half of all binding sites are occupied at equilibrium.
- is therefore equivalent to the molar concentration of ligand needed to half-saturate the protein.
- A lower = higher affinity (the protein binds the ligand more tightly, needing less of it to reach half-saturation).
- A higher = lower affinity.
Worked Example
Two proteins, A and B, bind the same ligand. Protein A reaches at . Protein B reaches at .
- ,
- Protein A has higher affinity because it is half-saturated at a lower ligand concentration.
Representative Values in Biology
| Protein | Ligand | (M) |
|---|---|---|
| Avidin (egg white) | Biotin | |
| Insulin receptor (human) | Insulin | |
| Anti-HIV immunoglobulin (human) | gp41 (HIV-1 surface protein) | |
| Nickel-binding protein (E. coli) | ||
| Calmodulin (rat) | to |
As a general reference for biological interactions: biotin–avidin is among the tightest known non-covalent interactions ( M), while enzyme–substrate interactions are relatively weak ( to M).
Part 3: The Heme Group
Both myoglobin and hemoglobin rely on the same non-protein component for oxygen binding: the heme group.
Structure of Heme
Heme is a prosthetic group — a non-protein molecule permanently or tightly associated with a protein to carry out its function. It consists of:
-
A heterocyclic porphyrin ring system: four pyrrole rings linked together by methine bridges, forming a large, flat, aromatic ring. The porphyrin ring carries various substituents including methyl groups, vinyl groups, and two propionate (negatively charged) side chains.
-
A single iron atom in the ferrous () oxidation state coordinated at the center of the porphyrin.
Iron Coordination Chemistry
Iron () forms six coordination bonds in total:
- Four bonds are in the plane of the porphyrin ring, each to a nitrogen atom from one of the four pyrrole rings.
- Two bonds are perpendicular to the plane of the porphyrin (one above, one below):
- The fifth coordination bond is occupied by the imidazole nitrogen of a histidine residue embedded in the protein — called the proximal histidine (His F8, i.e., the 8th residue of α-helix F; also designated His⁹³ in myoglobin’s sequence).
- The sixth coordination position is the oxygen-binding site.
Why Must Heme Be Inside a Protein?
Binding of to a free heme group (not embedded in a protein) is irreversible — the iron quickly oxidizes to (ferric iron) and loses the ability to bind oxygen reversibly. Enclosure within the hydrophobic protein pocket:
- Allows reversible oxygen binding.
- Protects iron from oxidation.
- Helps overcome the poor aqueous solubility of .
- Increases the effective diffusion of oxygen in tissue.
The Distal Histidine
In addition to the proximal His, there is a second key histidine — the distal histidine (His E7, also called His⁶⁴). This residue sits on the opposite side of the heme from the proximal His, near the oxygen-binding site. It forms a hydrogen bond with when it is bound, which:
- Stabilizes the bound
- Selectively increases the affinity for relative to other ligands like CO
Why Does Color Change?
When oxygen binds to heme, the electronic state of iron changes, causing a change in how the molecule absorbs visible light. This is why:
- Oxygenated (arterial) blood is bright red (oxyhemoglobin)
- Deoxygenated (venous) blood is deep purple-red (deoxyhemoglobin)
Heme and Carbon Monoxide Toxicity
Some small molecules bind to the heme iron with greater affinity than :
-
Carbon monoxide (CO): Binds with approximately 250 times greater affinity than . When CO occupies the sixth coordination site, is excluded. This is why CO is highly toxic to aerobic organisms — it effectively “locks” hemoglobin in a non-functional state. In physiological conditions, normal human blood contains ~1% CO-hemoglobin; in smokers, this rises to 8–10%. (Note: Fetal hemoglobin, HbF, has increased affinity for CO, making fetuses particularly vulnerable.)
-
Cyanide (): Binds tightly to methemoglobin (where iron is ), forming cyanmethemoglobin. It is then carried to tissues where it inhibits cytochrome c oxidase, the terminal enzyme of the mitochondrial electron transport chain, blocking ATP production.
-
Nitric oxide (NO): Binds to the of heme. NO is then released to the surrounding vascular cells during the transition from the R to T conformation of hemoglobin, producing a vasodilatory effect. Approximately 1 in every 1,000 hemoglobin molecules participates in this mechanism.
Part 4: Myoglobin
Identity and Location
Myoglobin is a small, monomeric, iron- and oxygen-binding globular protein found specifically in skeletal muscle and cardiac muscle (heart). Its structure was first solved by John Kendrew and Max Perutz in the period 1959–1968 — a landmark achievement in structural biology.
Primary Structure
Myoglobin is a single polypeptide of 153 amino acid residues with one heme group.
Secondary and Tertiary Structure
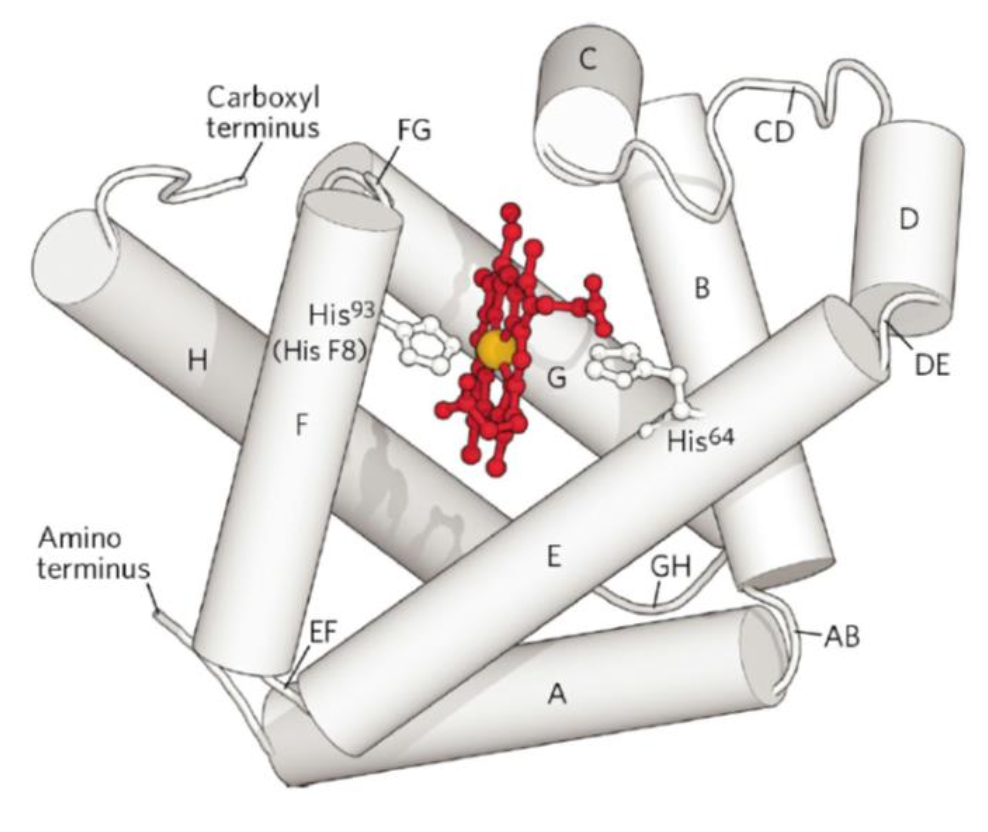
Myoglobin is a paradigmatic globin: a globular protein consisting almost entirely of α-helices. Specifically:
- 121 of 153 residues (78%) are found in α-helices.
- The structure contains eight α-helical segments, designated A through H.
- The helices are connected by short loop regions (bends), designated AB, CD, EF, FG, GH, etc., indicating which helices they connect.
- The heme group sits in a heme pocket primarily surrounded by helices E and F (helices B, C, G, and H also contribute nearby contacts).
The Role of the Proximal Histidine
The critical histidine that anchors the heme to the protein is:
- His⁹³ by sequence position (the 93rd amino acid from the N-terminus)
- His F8 by helix position (the 8th residue of helix F)
Its imidazole nitrogen forms the fifth coordination bond to .
The distal histidine (His⁶⁴, His E7) does not coordinate to iron directly, but hydrogen-bonds to bound .
When Oxygen Binds: Deoxymyoglobin vs. Oxymyoglobin
In the deoxygenated state (deoxymyoglobin), the iron atom sits slightly out of the plane of the porphyrin ring (displaced by ~0.4 Å toward the proximal His side), because the larger radius of deoxygenated doesn’t fit perfectly in the ring.
When binds, it occupies the sixth coordination position. The iron then adopts a smaller electronic radius and moves into the plane of the porphyrin — a ~0.5 Å shift. This pulls the proximal His (and the entire F helix) slightly along with it, a movement that has profound consequences in the multisubunit hemoglobin (discussed later).
Part 5: Oxygen Binding to Myoglobin — Quantitative Description
The Binding Equation for
Since the concentration of a dissolved gas is proportional to its partial pressure, we substitute partial pressure for concentration. Defining as the partial pressure at which half the binding sites are occupied (equivalent to ):
For myoglobin, (about 130 Pa).
Shape of the Binding Curve
The oxygen-binding curve of myoglobin is hyperbolic (not linear). This is characteristic of a single binding site with simple 1:1 stoichiometry. The curve rises steeply at low and then plateaus as the protein approaches saturation.
Physiological Interpretation
The affinity of myoglobin for oxygen is very high. In physiological conditions:
- At (resting muscle), myoglobin is approximately 95–98% saturated.
- Myoglobin’s affinity is so high that it barely changes across the range of arterial () to venous () blood oxygen pressures.
This means myoglobin readily accepts and holds oxygen over this range, functioning as an oxygen storage protein in muscle. Only when drops to very low levels — as during intense exercise — does myoglobin release its oxygen to the mitochondria.
Why Myoglobin Alone Can’t Serve as a Blood Oxygen Carrier
Precisely because myoglobin’s affinity is so high, it would be ineffective as an oxygen transport protein:
- It would load up with efficiently in the lungs ✓
- But it would never release sufficient in the tissues at physiological oxygen pressures ✗
The body needs a protein that releases oxygen in tissues while re-loading efficiently in the lungs. That role is filled by hemoglobin, which uses an elegant cooperative mechanism.
Part 6: Hemoglobin — Structure
Overview
Hemoglobin (abbreviated Hb) is the oxygen transport protein found in red blood cells (erythrocytes). Its key structural data:
- Molecular weight: 64,500 Da (~64.5 kDa)
- Tetrameric quaternary structure: four polypeptide chains, each carrying one heme group
- Adult hemoglobin (HbA): consists of two α chains and two β chains → designated
- α chains: 141 amino acid residues
- β chains: 146 amino acid residues
- α and β chains are 44% identical in sequence
- Each of the four subunits is structurally similar to myoglobin (all α-helical globin fold)
- The four heme groups are positioned near the surface, allowing efficient access to
- The quaternary structure forms from two identical dimers: and , assembling into
Extensive non-covalent contacts stabilize the four subunits, including hydrophobic interactions, hydrogen bonds, and ionic interactions (salt bridges).
Erythrocytes: The Container for Hemoglobin
Hemoglobin is carried in erythrocytes (red blood cells), where it constitutes approximately 34% of the cell by weight — each cell contains roughly 300 million hemoglobin molecules. A 70 kg adult carries approximately 750 g of hemoglobin.
During maturation from precursor cells (hemocytoblasts), developing red blood cells:
- Synthesize enormous quantities of hemoglobin
- Then lose all their organelles (nuclei, mitochondria, endoplasmic reticulum)
This makes mature erythrocytes essentially hemoglobin-filled sacs. They cannot replicate and survive for approximately 120 days in humans.
Because has only limited solubility in water ( M), hemoglobin within erythrocytes allows the blood to carry up to 68 times more oxygen than could be dissolved in plasma alone.
Arterial vs. Venous Saturation
- In arterial blood (from lungs to tissues): hemoglobin is about 96% saturated with
- In venous blood (from tissues back to the heart): hemoglobin is about 64% saturated
This difference — about 32% of total capacity — represents the oxygen actually delivered to tissues per circulation cycle.
Part 7: Cooperative Oxygen Binding by Hemoglobin
The Problem Hemoglobin Must Solve
Hemoglobin must:
- Load efficiently in the lungs, where (~100 mmHg)
- Unload efficiently in the tissues, where (~30 mmHg)
A protein with high affinity (like myoglobin) would load well but not unload. A protein with low affinity would unload well but not load. Neither alone solves the problem.
Hemoglobin solves this dilemma with cooperative binding: as each molecule binds, the remaining binding sites become progressively more receptive to additional molecules. This produces a sigmoid (S-shaped) binding curve — the hallmark of cooperativity.
The T State and R State
Hemoglobin exists in two distinct conformational states:
- T state (“tense”): Low affinity for . Stabilized by a large number of ionic interactions (salt bridges), especially at the (and ) interfaces. This is the deoxy conformation.
- R state (“relaxed”): High affinity for . Fewer stabilizing ion pairs; more “open” structure. This is the oxy conformation.
Oxygen binds to hemoglobin in either state but has significantly greater affinity in the R state. The protein begins predominantly in the T state when unloaded, and progressively shifts toward the R state as molecules bind.
The Sigmoid Binding Curve: A Physiological Advantage
The sigmoid curve means:
- At low (tissues): Hb is in the T state, has low affinity, and readily releases → efficient oxygen delivery
- At high (lungs): Hb transitions to the R state, has high affinity, and loads efficiently → effective pickup
This behavior is far superior to a simple hyperbolic curve (like myoglobin) for the purpose of oxygen transport.
Part 8: Molecular Mechanism of Cooperativity
Step 1 — Oxygen Binding Moves the Iron
In the deoxy (T) state, the iron atom sits slightly out of the plane of the porphyrin ring (displaced by ~0.4 Å toward the proximal histidine side). The porphyrin ring is slightly puckered.
When binds, it coordinates to the sixth position of . The iron is pulled into the plane of the ring (a shift of ~0.54 Å, or ~0.05 nm). The porphyrin ring flattens.
Step 2 — The Proximal Histidine and Helix F Move
Because His F8 (the proximal histidine) is covalently bonded to the protein backbone in helix F, the ~0.5 Å movement of iron pulls the proximal His, and hence the entire F helix, along with it.
Step 3 — Quaternary Structural Changes Propagate
The displacement of helix F alters the network of electrostatic interactions (salt bridges and ionic contacts) between subunits, particularly across the interface. Some of the ion pairs that stabilize the T state are broken. Some new ones form. The two dimers shift relative to each other by rotating approximately 15°, transitioning from the T to the R conformation.
This conformational change in one subunit communicates increased oxygen affinity to adjacent subunits, making it easier for them to bind in turn.
Summary of the Cooperativity Mechanism
- The first molecule binds weakly to a T-state subunit. Its binding energy is “spent” partly on loosening the intersubunit contacts.
- This binding triggers partial T→R transition, making subsequent binding easier.
- The last (fourth) molecule binds to a subunit already fully in the R state, with the highest affinity.
This is why the Hill coefficient for hemoglobin (, where means no cooperativity and would be perfect cooperativity) reflects strong but not absolute cooperativity.
Part 9: Two Theoretical Models of Cooperative Binding
Two major theoretical frameworks describe cooperative ligand binding in multisubunit proteins.
The Concerted Model (MWC Model)
Proposed by Monod, Wyman, and Changeux (1965)
Key assumptions:
- All subunits of the protein exist in one of two conformational states (T or R) simultaneously — no mixed states are allowed.
- In the absence of ligand, the T state is strongly favored.
- can bind to either state, but binds much more tightly to R.
- As ligand accumulates, the equilibrium shifts toward the R state for all subunits at once (hence “concerted” or “all-or-none”).
This model captures the overall sigmoid cooperative behavior remarkably well.
The Sequential Model
Proposed by Koshland and colleagues (1966)
Key assumptions:
- Ligand binding induces a conformational change in the individual subunit it binds to.
- That local conformational change influences neighboring subunits, making their conformational change (and ligand binding) more likely.
- Mixed states of subunits are permitted — not all or none.
Reconciling the Two Models
The two models are not mutually exclusive:
- The concerted model can be viewed as the extreme “all-or-none” limiting case of the sequential model.
- Real hemoglobin cooperativity likely involves elements of both — essentially a concerted transition at the dimer level, with sequential features at the tetramer level.
Part 10: The Bohr Effect — Transport of and
Hemoglobin does far more than transport oxygen. It also plays a major role in transporting the waste products of metabolism — and — from tissues back to the lungs.
The Bohr Effect: Defined
The Bohr effect refers to the inverse relationship between concentration (i.e., pH) and hemoglobin’s affinity for oxygen:
- High (lungs) → Hb binds → releases
- Low (tissues) → Hb releases → binds
In other words:
- stabilizes the T state (deoxygenated, low affinity)
- Oxygenation releases (T→R transition disrupts ion pairs that held protons)
Molecular Basis: His 146
About 50% of the Bohr effect is caused by changes in the pKa of His 146 of the β subunits (His HC3). In the T state, this residue is protonated () and forms a stabilizing salt bridge to Asp 94 (Asp FG1) on the same chain. When hemoglobin shifts to the R state upon binding, this salt bridge is disrupted, and the proton on His 146 readily dissociates. Additional residues (including the amino-terminal residues of α subunits, and other His residues) contribute similarly.
Important clarification: oxygen binds to the heme iron, while binds to amino acid residues in the protein — these are distinct sites, but they influence each other allosterically.
Physiological Significance of the Bohr Effect
In the peripheral tissues, metabolic activity generates and . This increased acidity shifts the oxygen-binding curve to the right (lower affinity), promoting oxygen release exactly where it is needed. The overall effect is physiologically elegant:
- Hemoglobin picks up protons and releases in metabolically active tissues.
- In the lungs, binding displaces , which is then used to regenerate for exhalation.
Hemoglobin buffers approximately 30–40% of the acid produced by tissues.
Transport by Hemoglobin
In the tissues, produced by cellular metabolism enters erythrocytes and:
-
(~70–80%) Is hydrated to bicarbonate by carbonic anhydrase:
The is exported to the plasma, and the is buffered by hemoglobin (Bohr effect). -
(~15–20%) Reacts directly with the α-amino groups at the N-termini of globin chains to form carbamate (carbaminohemoglobin):
The carbamate groups also form additional salt bridges that help stabilize the T state (further promoting release), and the reaction releases that also contributes to the Bohr effect.
Hemoglobin is responsible for transporting approximately 15–20% of total and buffering approximately 40% of total produced by tissues.
In the lungs, the process reverses:
- binds → T→R transition → carbamate and proton release
- Released combines with → →
- is exhaled
Effects of pH and Temperature on the Binding Curve
As a consequence of the Bohr effect:
- Decreasing pH (more acidic, as in active tissues) shifts the oxygen-binding curve to the right → lower affinity, more released
- Increasing pH shifts the curve left → higher affinity
Temperature also affects binding:
- Lower temperatures (e.g., 10°C) shift the curve left → higher affinity (less delivered to tissues)
- Higher temperatures (e.g., 43°C, as in fever or exercising limbs) shift the curve right → lower affinity, more released where metabolic demand is greatest
Part 11: Allosteric Regulation — 2,3-Bisphosphoglycerate (BPG)
What Are Allosteric Proteins?
An allosteric protein is one in which binding of a molecule at one site influences the binding properties of a different site on the same protein. Hemoglobin is the classic example.
Key terminology:
- Modulator: the molecule that binds to the allosteric (regulatory) site
- Homotropic: when the normal ligand (here, ) is also the modulator — cooperative binding of to Hb is a homotropic interaction
- Heterotropic: when the modulator is a different molecule from the main ligand — BPG, , and are heterotropic modulators of hemoglobin
2,3-Bisphosphoglycerate (BPG): Structure and Origin
2,3-Bisphosphoglycerate (BPG) is a highly negatively charged metabolite ( negative charges at physiological pH) produced in erythrocytes as a side product of glycolysis:
(where DPGM = diphosphoglycerate mutase, DPGP = diphosphoglycerate phosphatase)
BPG is present in erythrocytes at high concentrations — comparable to that of hemoglobin itself.
How BPG Regulates Hemoglobin
BPG greatly reduces hemoglobin’s affinity for oxygen. One molecule of BPG binds to each hemoglobin tetramer. Its binding site is the central cavity between the two β subunits — but only in the T (deoxy) state. BPG’s multiple negative charges form electrostatic interactions with positively charged groups lining this cavity (from both β subunits).
When Hb transitions to the R state, the central cavity narrows and can no longer accommodate BPG. BPG is therefore expelled, and the R state is free of BPG.
Net effect:
- BPG stabilizes the T state → decreases affinity → promotes release in tissues
- In the absence of BPG, hemoglobin converts more easily to the R state and has much higher affinity (closer to that of myoglobin in terms of the curve shape)
This provides a crucial regulatory mechanism for tuning oxygen delivery to match physiological needs.
BPG and Altitude Adaptation
At high altitude, the atmospheric is lower, making it harder for hemoglobin to load oxygen in the lungs and harder to deliver adequate oxygen to tissues.
Adaptation mechanism:
- Initially: delivery at high altitude drops to ~30% of maximum
- Within hours to days: erythrocytes increase BPG production in response to hypoxia
- The BPG concentration in blood rises from ~5 mM (sea level) to ~8 mM (high altitude)
- The increased BPG lowers Hb’s affinity, causing the binding curve to shift right
- Now, despite lower in the lungs, hemoglobin still loads and — more importantly — delivers ~37% of its capacity to tissues, approximately restoring normal oxygen delivery
This is a beautiful example of metabolic adaptation.
Part 12: Fetal Hemoglobin (HbF)
A Special Problem: Oxygen Transfer from Mother to Fetus
The developing fetus must acquire oxygen from its mother’s blood across the placenta. Maternal hemoglobin must release at the placenta for fetal hemoglobin to pick it up. For this to work, fetal hemoglobin must have higher affinity for than maternal (adult) hemoglobin.
Structure of HbF
Fetal hemoglobin (HbF) has the composition :
- α subunits are identical to adult α chains (141 residues)
- γ subunits replace the β chains (146 residues; 39 amino acids differ from the β chain)
Why HbF Has Higher Affinity
A critical structural difference: the γ subunit of HbF cannot bind BPG effectively. Because BPG normally decreases affinity, the absence of BPG binding means HbF has intrinsically higher affinity than adult HbA.
At the placenta:
- Maternal Hb (HbA, lower affinity) releases
- Fetal Hb (HbF, higher affinity) captures that
This affinity difference is visible in the -saturation curves: at any given , HbF is more saturated than HbA, and the curve lies to the left of the adult curve.
Part 13: Sickle Cell Anemia — A Single Mutation with Catastrophic Consequences
The Genetic Defect
Sickle cell anemia is caused by a single point mutation in the gene encoding the β subunit of hemoglobin:
Glutamate (Glu) → Valine (Val) at position 6 of the β chain
This produces hemoglobin S (HbS) instead of normal hemoglobin A (HbA):
- HbA β chain begins: Val·His·Leu·Thr·Pro·Glu·Glu·Lys…
- HbS β chain begins: Val·His·Leu·Thr·Pro·Val·Glu·Lys…
Why This One Change Is So Damaging
Glutamate carries a negative charge at physiological pH; valine is nonpolar and hydrophobic. The substitution:
- Removes a negative charge from the surface of the β chain
- Creates a “sticky” hydrophobic patch on the exterior of hemoglobin
In the oxygenated (R) state, this patch is hidden — HbS behaves relatively normally. However, in the deoxygenated (T) state, this hydrophobic Val residue on one HbS tetramer fits into a complementary hydrophobic pocket on an adjacent deoxy-HbS tetramer. This intermolecular contact promotes the formation of long, insoluble fibrous polymers (tubular fibers) of deoxygenated HbS molecules.
Consequences of Polymerization
- The insoluble fibers distort the normally flexible biconcave disc-shaped erythrocytes into a rigid sickle (crescent) shape
- Sickled cells are stiff and cannot easily navigate small capillaries
- They obstruct blood flow in capillaries → ischemia (tissue oxygen deprivation), severe pain, and organ damage
- Sickled cells are also more fragile and shorter-lived → hemolytic anemia (destruction of red blood cells)
Inheritance Pattern
Sickle cell anemia is an autosomal recessive disease:
- Homozygous (HbS/HbS): severe disease; untreated, patients often die during childhood; with modern treatment, median life expectancy is 42–47 years
- Heterozygous (HbA/HbS, “sickle cell trait”): generally healthy at normal activity levels; can experience symptoms during intense exercise or at high altitude due to reduced
Clinical Manifestations
- Severe pain episodes (vaso-occlusive crises)
- Stroke
- Pulmonary and heart disease
- Fatigue and decreased hemoglobin (anemia)
- Eye damage
- Bacterial infections (immune dysfunction)
- Leg ulcers
- Thrombosis in the spleen and liver
- Swelling and inflammation of fingers/toes, arthritis-like symptoms
A Striking Irony: Protection Against Malaria
The HbS allele is maintained at high frequency in malaria-endemic regions (sub-Saharan Africa, parts of the Mediterranean, Middle East, and India) because heterozygous HbA/HbS individuals are partially protected against severe malaria caused by Plasmodium falciparum. The malaria parasite has difficulty surviving and reproducing in sickled or sickling-prone erythrocytes.
Current Treatments
| Treatment | Mechanism |
|---|---|
| Hydroxyurea | Increases fetal hemoglobin (HbF) production; reduces frequency of pain crises and transfusion need |
| Voxelotor | Increases red blood cell flexibility (stabilizes HbS in oxy form); reduces anemia |
| Crizanlizumab | Monoclonal antibody; prevents sickled cells from clumping (reduces pain crises) |
| Painkillers (acetaminophen, ibuprofen, opioids) | Manage pain during crises |
| Antibiotics | Prevent infections (especially daily penicillin in young children) |
| Blood transfusions | Reduce stroke risk and treat severe anemia |
| Bone marrow / stem cell transplant | Only potential cure; limited by donor availability and significant procedural risk |
Summary: Myoglobin vs. Hemoglobin Compared
| Property | Myoglobin (Mb) | Hemoglobin (Hb) |
|---|---|---|
| Location | Skeletal muscle, heart | Erythrocytes (blood) |
| Subunit composition | Monomer (1 chain) | Tetramer () |
| Heme groups | 1 | 4 |
| Quaternary structure | None | |
| Binding curve shape | Hyperbolic | Sigmoid (cooperative) |
| ~2.8 mmHg (very low) | ~26 mmHg (physiological range) | |
| Bohr effect | No | Yes |
| BPG sensitivity | No | Yes |
| Cooperativity | No | Yes (homotropic) |
| Primary function | storage in muscle | transport from lungs to tissues; and transport back |
Key Equations to Remember